 手机版
手机版 化工仪器网手机版
化工仪器网手机版
 化工仪器网小程序
化工仪器网小程序
 官方微信
官方微信 公众号:chem17
公众号:chem17
 扫码关注视频号
扫码关注视频号











一、NALP3炎性体的结构和分布
NALP3炎性体是一类分子量约为700kDa的大分子蛋白复合体,由核苷酸结合寡聚化结构域样受体(nucleotide-binding oligomerization domain-like receptors,NLRs)家族成员NALP3、衔接蛋白ASC(apoptosis-associated speck-like protein containing a CARD)和CARDINAL,以及效应蛋白caspase-1组成,在细胞质内发挥外源性微生物或内源性危险信号感受器的作用,是活化半胱天冬酶-1(caspase-1)的分子平台,调控IL-1β、IL-18、IL-33等促炎细胞因子的成熟和分泌,在天然免疫、获得性 免疫反应中发挥重要作用。研究表明,激活NALP3炎性体的内源性危险信号包括ATP、尿酸晶体、活性氧(reactive oxygen species,ROS)、β淀粉样蛋白、细胞外基质成分和溶酶体酶等。粒细胞、单核细胞、树突细胞、B细胞和T细胞都能产生NALP3, 但它主要分布在口咽、食管、宫颈阴道粘膜的非角质化上皮、成骨细胞,膀胱和输尿管的上皮细胞也能表达NALP3。NALP3炎性体在2型糖尿病、痛风、阿尔兹海默病和肾脏疾病等非感染性炎症疾病中的作用和意义日益受到关注。
二、NALP3炎性体的活化及信号通路
微生物毒素:如尼日利亚菌素、气单、胞菌溶素、刺尾鱼毒素等或T3SS和T4SS的转位 元件通过介导微孔结构活化NALP3炎性体。有趣的是,ATP-P2X7R活化的缝隙连接蛋白pannexin-1也能介导细胞膜的微孔结构,使微生物分子(如胞壁酰二肽)进入细胞质,被NALP3识别,继而活化炎性体。
胞外ATP激活嘌呤型P2X7受体非选择性阳离子通道,随后pannexin-1通道逐渐开放,导致钾离子外流,钙离子内流,zui后活化NALP3炎性体。ATP可以从损伤的细胞和细胞应激中释放,内皮细胞和上皮细胞的机械性刺激也能释放ATP。zui近研究发现,治疗2型糖尿病的药物格列苯脲作用于P2X7受体下游,通过和KATP通道结合,阻止钾离子外流从而抑制炎性体的活化。值得注意的是,毒素介导的微孔结构以及晶体物质(如尿酸钠 晶体)不能被细胞吞噬也能引起钾离子外流,导致NALP3炎性体的活化。
激活NALP3炎性体的信号通过诱导活性氧的产生,引起未识别蛋白的构像变化从而活化NALP3炎性体。所有已知的NALP3炎性体的活化信号(石棉、二氧化硅、尿酸钠晶体等)都 能诱导ROS产生,ROS抑制剂可以阻断炎性体的活化。如果细胞吞噬功能障碍或丧失使晶体物质 不能被细胞吞噬消化(frustrated phagocytosis),可以 通过激活尼克酰胺腺嘌呤磷酸二核苷酸(NADPH)氧化酶催化反应产生大量活性氧自由基,继而活化NALP3炎性体。
微粒样物质如尿酸钠晶体、二氧化硅或β淀粉样蛋白被细胞吞噬后,破裂的溶酶体释放组织蛋白酶-B,剪切未知的底物从而活化炎性体。溶酶体损伤释放组织蛋白酶-B是NALP3炎性体活化zui重要的上游信号通路。
三、NALP3炎性体对非感染性炎症疾病的作用
(一)、NALP3炎性体与2型糖尿病2型糖尿病:
IL-1β等致炎因子通过对抗胰岛素信号促进胰岛素抵抗,抑制胰岛素依赖的葡萄糖摄取,降低糖耐量;同时,分泌胰岛素的胰岛局部炎症也参与了糖尿病的发展,伴随免疫细胞的浸润、局部细胞炎症因子的升高,局部的炎症和葡萄糖的毒性效应引起β细胞的凋亡。长期的高血糖抑制β细胞分泌胰岛素,诱导IL-1β依赖方式的细胞死亡。糖尿病病人或动物模型的胰岛β细胞代谢应激时表达IL-1β,相比巨噬细胞分泌较少的IL-1β在抑制β细胞活性和胰岛素分泌能力是足够的。因此,有学者提出IL-1β是2型糖尿病发病的重要驱动因素。新近研究表明,NALP3炎性体是活化IL-1β的分子平台,NALP3炎性体在2型糖尿病发生发展中的作用日益受到关注。Zhou等用酵母双杂交技术已证实硫氧环蛋白相互作用蛋白(thioredoxin-interacting protein,TXNIP)是NALP3结合蛋白,在高糖环境下,TXNIP-/-和NALP3-/-小鼠胰岛β细胞IL-1β的水平显著低于野生型小鼠。进一步研究发现,ROS抑制剂吡咯烷二硫氨基甲酸盐(APDC)或NADPH氧化酶抑制剂能明显抑制高糖介导的NALP3炎性体的活性、降低IL-1β水平。Schroder等认为,持续高糖不仅能诱导胰岛β细胞表达大量TXNIP,而且能通过上调NADPH氧化酶或线粒体损伤产生ROS,使TXNIP从硫氧还蛋白(thioredoxin,TRX)中分离出来, 产生的TXNIP直接活化NALP3炎性体,引起IL-1β的成熟和释放,活化的IL-1β一方面直接引起胰岛β细胞的死亡和损伤, 另一方面通过炎症反应和免疫细胞浸润进一步加重胰岛β细胞功 能障碍,zui终导致2型糖尿病。这些研究结果提示,阻止NALP3炎性体的活化可以成为预防和治疗2型糖尿病的手段。
(二)、NALP3炎性体与痛风
痛风是普通的自发炎症性关节炎,以血清高尿酸和反复发作的关节内尿酸单钠结晶沉积为特征,成年人发病率约1~2%。zui近研究认为,IL-1β是介导痛风性关节炎的重要炎症因子,IL-1β在痛风急性发作中发挥关键作用。IL-1β的成熟依赖caspase-1,NALP3炎性体是活化caspase-1的分子平台。Martinon等用尿酸钠晶体(monosodiumurate,MSU)刺激腹膜巨噬细胞的体内和体外实验均发现,NALP3-/-、ASC-/-以及Caspase-1-/-小鼠腹膜炎症反应比野生型小鼠显著减轻,IL-1β的成熟和释放受到明显抑制。说明NALP3炎性体在尿酸钠晶体介导IL-1β,引起痛风性炎症反应中扮演重要角色。
进一步研究发现,秋水仙碱通过抑制MSU被细胞吞噬,进而抑制NALP3炎性体的活化,阻止IL-1β的成熟来治疗痛风急性发作。另有研究用MSU刺激THP-1(人单核细胞株)和原代小鼠巨噬细胞发现,当细胞外高钾(Kcl浓度130mM)时,钾离子外流受到抑制,也阻止了NALP3炎性体的活化,细胞培养液上清和细胞裂解液中活化的IL-1β蛋白表达明显降低。而NALP3 -/- 的细胞无论胞外高钾(Kcl浓度130mM)或正常钾离子浓度,MSU刺激 后,细胞培养液上清和细胞裂解液中几乎没有活化的IL-1β。此外,ROS抑制剂N-乙酰半胱氨酸(NAC)预处理THP-1细胞,MSU刺激后,IL-1β的成熟也明显受到抑制。
痛风是普通的自发炎症性关节炎,以血清高尿酸和反复发作的关节内尿酸单钠结晶沉积为特征,成年人发病率约1~2%。zui近研究认为,IL-1β是介导痛风性关节炎的重要炎症因子,IL-1β在痛风急性发作中发挥关键作用。IL-1β的成熟依赖caspase-1,NALP3炎性体是活化caspase-1的分子平台。Martinon等用尿酸钠晶体(MSU)刺激腹膜巨噬细胞的体内和体外实验均发现,NALP3-/-、ASC-/-以及Caspase-1-/-小鼠腹膜炎症反应比野生型小鼠显著减轻,IL-1β的成熟和释放受到明显抑制。说明NALP3炎性体在尿酸钠晶体介导IL-1β,引起痛风性炎症反应中扮演重要角色。
(三)、NALP3炎性体与阿尔兹海默病
IL-1β在阿尔兹海默病的发病过程中起关键作用,β淀粉样蛋白(amyloid-β)诱导小神经胶质细胞产生大量IL-1β,诱发脑内炎症反应,加重脑组织的损伤。NALP3炎性体参与调控IL-1β的活化和分泌。Halle等的研究结果显示,细胞外Aβ被原代小鼠小神经胶质细胞吞噬后导致溶酶体 损伤,释放组织蛋白酶B,激活NALP3炎性体,活化caspase-1,zui终产生和释放促炎细胞因子IL-β,引起脑组织的炎症反应和神经元损伤。提示Aβ通过激活NALP3炎性体,使IL-1β成熟和释放,引起脑组织的炎症反应可能是阿尔兹海默病的重要机制。
(四)、NALP3炎性体与肾脏疾病
NALP3炎性体在肾脏疾病中的作用目前受到广泛关注。Iyer等通过肾脏缺血-再灌注(I/R)模型,分别观察野生型小鼠、NALP3-/-和ASC-/-小鼠肾功能、肾小管损伤、肾组织炎症反应以及小鼠死亡率等指标的变化,发现野生型小鼠NALP3和ASC mRNA表达显著增加并伴有严重肾小管坏死,细胞外基质双糖链蛋白聚糖及透明质酸的沉积显著增加,肾间质中性粒细胞浸润明显增多,肾脏IL-1β和中性粒细胞化学趋化因子KC水平显著上升。
与之相反,NALP3-/-和ASC-/-小鼠肾组织炎症反应和肾功能损伤明显减轻,小鼠死亡率显著降低。说明NALP3炎性体在非免疫介导的间质性肾损伤中具有重要的作用。新近研究发现,在单侧输尿管梗阻(unilateral ureteral obstruction,UUO)模型中,Biglycan-/-小鼠肾脏caspase-1活性和IL-1β水平显著下降肾间质浸润的单核细胞数量减少以及肾小管损伤评分显著降低;而在野生型小鼠,双糖链蛋白聚糖大量聚集于肾小管上皮细胞,先于肾间质巨噬细胞的浸润,肾脏caspase-1活性、IL-1β水平明显增高。进一步说明非免疫介导的间质性肾损伤可能的机制是通过双糖链蛋白聚糖活化NALP3炎性体,激活caspase-1,剪切无活性的IL-1β前体,zui后分泌和释放成熟IL-1β,导致急性间质性肾损伤。上述研究结果提示,NALP3炎性体参与了急性间质性肾损伤的发生发展,切断其活化环节能有效地防止肾组织炎症反应,为预防和治疗急性肾损伤炎症反应提供了的新的思路。
四、结语
NALP3炎性体不仅在机体对抗病原微生物的天然免疫中发挥重要作用,也能通过识别内源性危险信号、代谢性应激介导获得性免疫反应。NALP3炎性体活化在非感染性炎症疾病的急、慢性炎症反应等病理生理过程中发挥重要的作用和意义。初步的研究结果已证实NALP3炎性体参与了2型糖尿病、痛风、肾脏疾病等急慢性非感染性炎症疾病的发生发展,阻止NALP3炎性体的活化可能成为防治这些疾病的一个新的靶点,但还需要进一步研究来阐明其作用机制,为其临床运用提供理论依据。
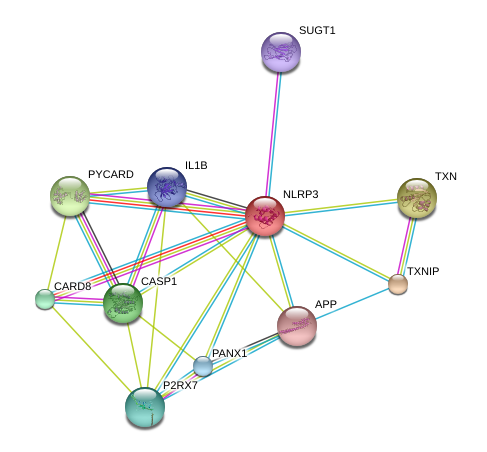
相关通路指标:
Innate Immunity: CASP1 (ICE), CHUK (IKKα), IKBKB, IRS1, IRS2, NLRP3, NFKBIA (IκBα/MAD3), PYCARD (TMS1/ASC), RELA, TLR4.
Inflammation: ALOX5, CASP1 (ICE), CCL2 (MCP-1), CCR4, CCR5, CHUK (IKKα), CXCR4, IFNG, IKBKB, IL1B, IL23R, IL6, IL8, LTA4H, NLRP3, OLR1, PYCARD (TMS1/ASC), RELA, TNF, TNFRSF1A, TNFRSF1B.
Apoptosis: PPARG, SERPINE1 (PAI-1), TNF, CASP1 (ICE), IKBKB, IRS2, MAPK9 (JNK2), NFKBIA (IκBα/MAD3),NLRP3, PYCARD (TMS1/ASC), RELA, TNFRSF1A, JAK2, PIK3CA (p110A), SOCS3, RPS6KB1, TNFRSF1B.
Defense Response to Viruses: CD4, CD40 (TNFRSF5), CD86, CD8A, CXCL10 (INP10), DDX58 (RIG-I), HLA-A, IFNAR1, IFNB1, IL23A, IL6, IRF3, NLRP3, TICAM1 (TRIF), TLR3, TLR7, TLR8, TYK2.
Nod-Like Receptor Signaling:
Receptors and Signaling: AIM2, CARD9, CASP1 (ICE), HSP90AA1, MEFV, NLRP3, NOD2, OAS2, PSTPIP1, PYCARD (TMS1/ASC), PYDC1 (POP1), SUGT1.
Responsive Genes: IL1B, IL18.
通路研究工具:
抗菌响应系列 | PCR Array of Antibacterial Response | HU-ABR-084 |
抗真菌响应系列 | PCR Array of Antifungal Response | HU-AFR-084 |
抗病毒反应系列 | PCR Array of Antiviral Response | HU-AR-084 |
细胞凋亡系列 | PCR Array of Apoptosis | HU-APO-084 |
炎性体系列 | PCR Array of Inflammasomes | HU-IFM-084 |
胰岛素抵抗系列 | PCR Array of Insulin Resistance | HU-INR-084 |
先天和适应性免疫应答系列 | PCR Array of Innate & Adaptive Immune Responses | HU-IAIR-084 |
BioTNT PCR Array:
Q-PCR array是信号通路基础生物学研究和临床疾病研究的重要工具。这种功能分类芯片运用成熟的SYBR Green荧光定量PCR技术,专注于一个信号通路中基因的表达水平检测。与传统的高密度表达谱芯片相比,Q-PCR array具有针对性强、灵敏度高、准确可靠等优点。 芯片上的基因包括了与研究对象有确定关系的基因,或至少是与研究对象的关系有待考证的基因。如果研究对象是某一生物学通路的基因,则使用针对该类基因的功能分类基因芯片比使用高密度表搭配芯片更加有效便利。
BioTNT QPCR芯片特点:
l 灵敏度高,样本使用量低;
l 线性范围广,可同时检测表达水平差异大的基因;
l 每个基因的检测引物优化后保证高扩增率和产物单一;
l 重复性高,Ct值的平均差异只有0.25个循环;
BioTNT QPCR Array芯片操作流程: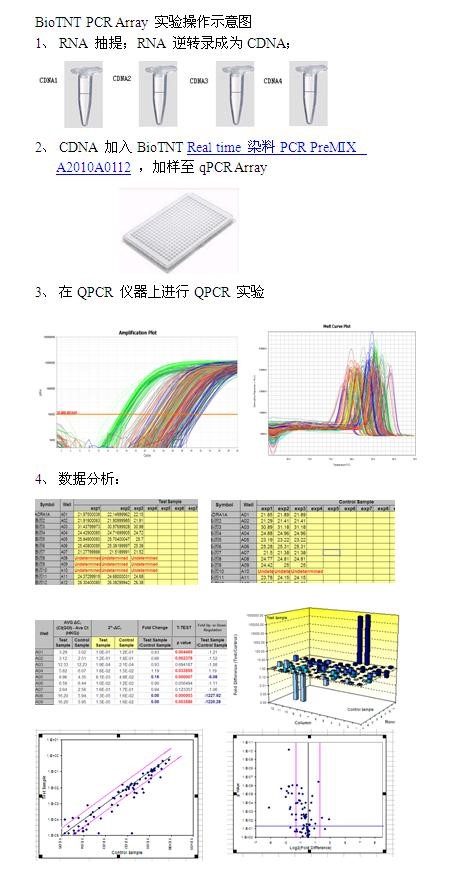
相关产品




免责声明
- 凡本网注明“来源:化工仪器网”的所有作品,均为浙江兴旺宝明通网络有限公司-化工仪器网合法拥有版权或有权使用的作品,未经本网授权不得转载、摘编或利用其它方式使用上述作品。已经本网授权使用作品的,应在授权范围内使用,并注明“来源:化工仪器网”。违反上述声明者,本网将追究其相关法律责任。
- 本网转载并注明自其他来源(非化工仪器网)的作品,目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责,不承担此类作品侵权行为的直接责任及连带责任。其他媒体、网站或个人从本网转载时,必须保留本网注明的作品第一来源,并自负版权等法律责任。
- 如涉及作品内容、版权等问题,请在作品发表之日起一周内与本网联系,否则视为放弃相关权利。




 采购中心
采购中心
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}